library(bio3d)
p <- read.pdb("hivpr_23119/hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb")Class 11: AlphaFold
Background
We saw last day that the main repository for biomolecular strucutre (the PDB database) only has ~250,000 entries
UniProtKB (the main protein sequence database) has over 200 million entries!
AlphaFold
In this hands-on session we will utilize AlphaFold to predict protein structure from sequence (Jumper et al. 2021).
Without the aid of such approaches, it can take years of expensive laboratory work to determine the structure of just one protein. With AlphaFold we can now accurately compute a typical protein structure in as little as ten minutes.
The EBI AlphaFold database
The EBI AlphaFold database contains lots of computed structure models. It is increasingly likely that the structure you are intrested in is already inn this database at https://alphafold.ebi.ac.uk/
There are 3 major outputs from AlphaFold
- A model of structure in PDB format.
- A pLDDT score: that tells us how confident the model is for a given residue in your protein (High values are good, above 70)
- A PAE score that tells us about protein packing quality.
If you can’t find a match entry for the sequence you are interested in AFDB you can run AlphaFold youself…
Running AlphaFold
We will use ColabFold to run AlphaFold on our sequence https://github.com/sokrypton/ColabFold
Figure from AlphaFold
![]()
Interpreting Results
Custom analysis of resulting models
We can read all the AlphaFold results into R and do more quantitative analysis than just viewing the structures in Mol-star:
Read all the PDB models:
pdb_files <- list.files("hivpr_23119/", pattern = ".pdb", full.names=T)
pdbs <- pdbaln(pdb_files, fit=TRUE, exefile="msa")Reading PDB files:
hivpr_23119/hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
hivpr_23119/hivpr_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
hivpr_23119/hivpr_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
hivpr_23119/hivpr_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
hivpr_23119/hivpr_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb
.....
Extracting sequences
pdb/seq: 1 name: hivpr_23119/hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
pdb/seq: 2 name: hivpr_23119/hivpr_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
pdb/seq: 3 name: hivpr_23119/hivpr_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
pdb/seq: 4 name: hivpr_23119/hivpr_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
pdb/seq: 5 name: hivpr_23119/hivpr_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb #library(bio3dview)
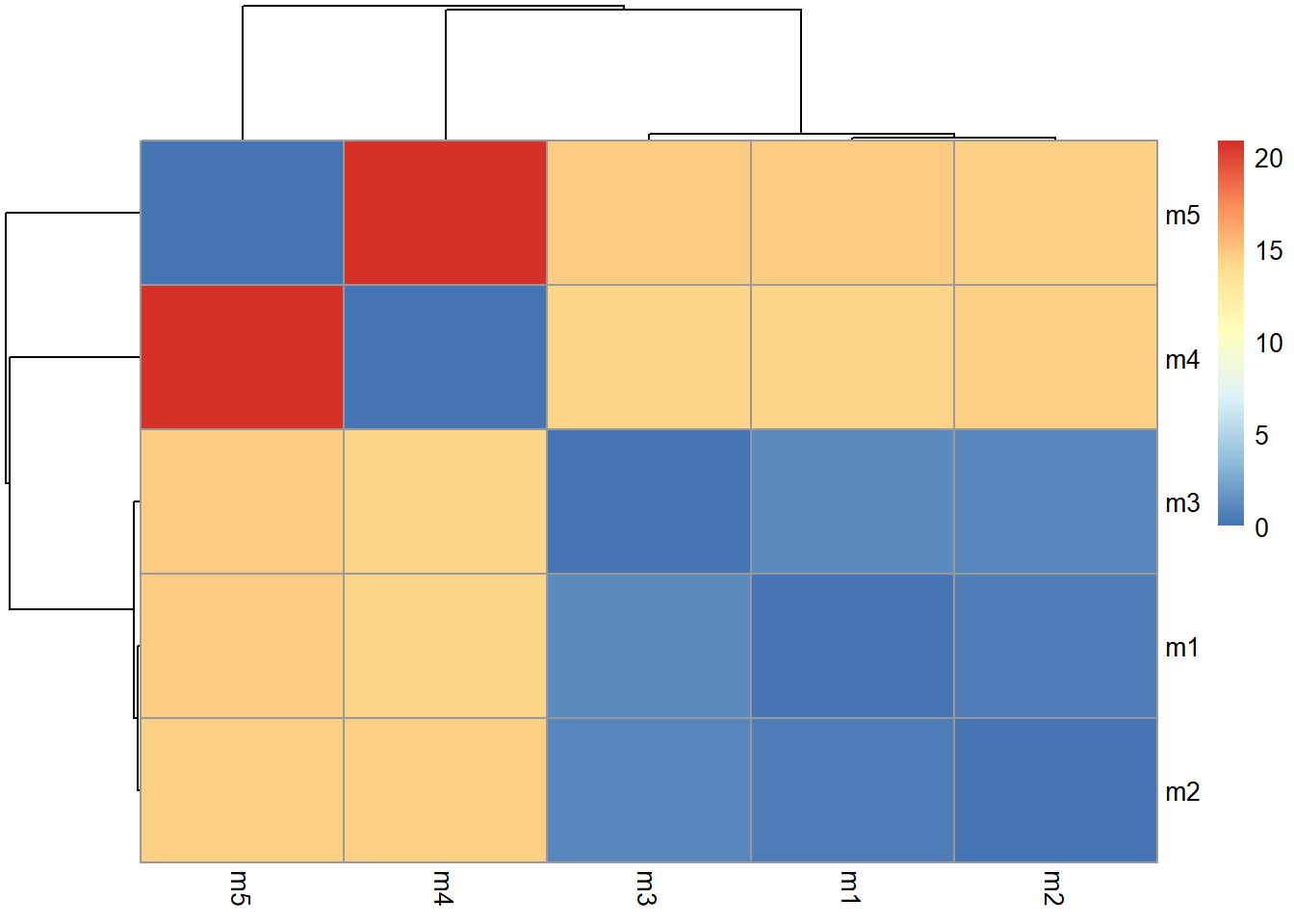
#view.pdbs(pdbs)How similar or different are my models?
rd <- rmsd(pdbs)Warning in rmsd(pdbs): No indices provided, using the 198 non NA positionslibrary(pheatmap)
colnames(rd) <- paste0("m",1:5)
rownames(rd) <- paste0("m",1:5)
pheatmap(rd)